User ManualPrimer designUser inputsDefault settingsResultiBP-seqGenotypingSubmit a taskResultEpigenotypingSubmit a taskResultCopyright
User Manual
Primer design
Primer design is divided into user input area and parameter setting area. To get primer design results, you need to submit at least the items in the user input area.
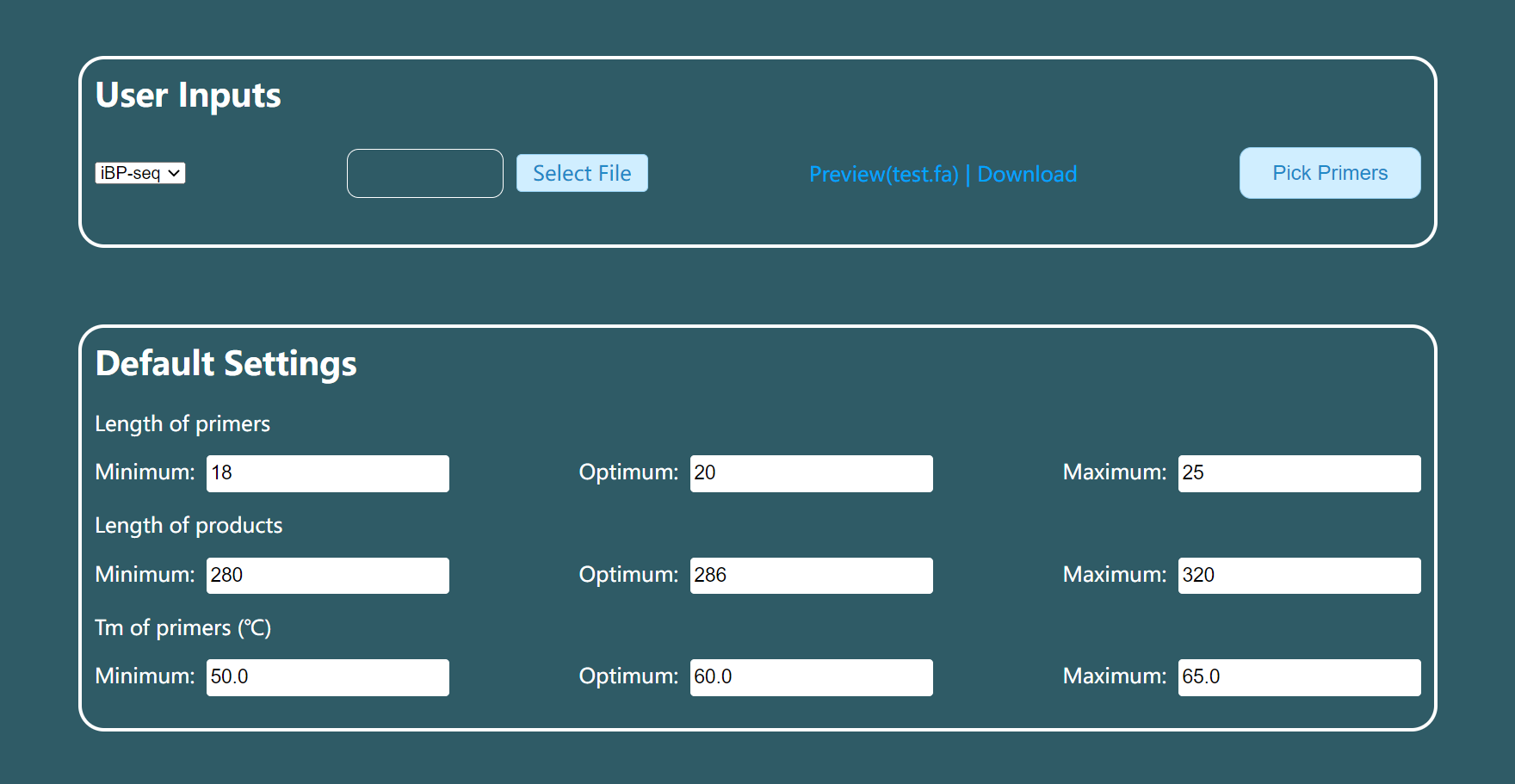
User inputs
[ required ] Template sequence file in fasta format
For the description line of each sequence in fasta (beginning with the > symbol), we use the
"|"symbol to separate different sequence attributes. The described sequence attributes include the name of the molecular marker and the position of the molecular marker. In addition, we recommend that each fasta sequence be longer than 600bp for better primer design results.Describe line template:
> marker_name | marker_areamarker_name The name of the molecular marker, it is recommended to use English letters and Arabic numerals, and avoid spaces and irregular characters.
marker_position To ensure the sequencing quality of the mutated sequence, the targeted sites were within 150nt of the forward or within 118nt of the reverse target-specific primers. The bases on the sequence are counted from 1, including two ways of writing:
- The position of the single nucleotide in the sequence. For example, 348 means that the 348th base on the next line of sequence is a molecular marker, and this expression is only applicable to single-nucleotide polymorphism (Single-Nucleotide Polymorphism, SNP).
- When the length of molecular markers is greater than or equal to 1 (such as InDel and so on), the start and end positions of the markers are separated by
"-"symbols. Such as 301-304, which means that the 4 bases in the range of 301 to 304 bases on the next line of sequence are molecular markers, and for SNP markers, there are also equivalent expressions, such as 348-348, which is consistent with the first way of writing. Therefore, this form of expression is suitable for most molecular markers.
Data Format
>marker1|301 TTTGCCACTTAAACGTTGGCTCCTTGACTGTGTCATGTGTCCATGTGTGTCGATTCTGAAAGATTCCTTCTCCTTGGGTGTTCAGTTTCTTGATTGATGAAAATTGCCTGGCTAACTAATCTGTGTTTCCTCGCCAGGCAATTGCGCTCTGCCCCGACGTCGCCGTCTACTGGATCAATCGGGCCCTGTGCCATTTCAAGCGCAAGTGAGCGACTTTACTTTTTGTTGCCTCCCTTCCTGGCAAGAAAGAAACAAACGTCTGGAATAGTAATCTTTGTGCACAGGGAGTGGGCCAAGGTCGAAGAGGACAGCAGGAGGGCTCTTGCGCTTGATTACACTTTAGTCAAGGTCACATTCTTGTTTCTTCTTTCGTCTCAAAACTAAAGCGCCGACTTCAGTTACCTCTACATTATGTAATCGCTATTCTTAACTAACCGTTTGACACCAATAAAGATACAAAAGTAAGTAGCCGTGATTATTCCGTTCCTTGTGTGATGTAGGGGCACTATCTGCTGGGGTGTGCGCTACTCGAGAAGGAAGAATCTGCTCTTGCTATCAAGGAATTCGAAAAGGTTTACCTCAACCGTTTGTCTCTTGCACG
>marker2|301-304TGCTCGGATGTGACTGAGTGACGACGATCACGGTGCAGTTTCTTGTCAATTCTTCGGGTCGAATGCTCGCAGCAGACGACTGAGGCCGTGATCAAGCGGTGGACGACGGTGACGGAGATCATAGGGAAGGCCATGGTCGCCATGCGGAGGCAGCTGTACGCGCAGAACCCAGGCGCCTTCGACGGTTTCAGGGACGAATACTTGCTGGCCATCGCGGAGAACCGCATCCTGATCCTGCTCGACTTCGCCAACGGATTCACCAGCATCACGTCGCACGAGAAGCTCGTGTACATGCTCGGCATGTACGAGGCTCTCACCGACGCCGCTCCCAGCCTCCTGCTCCTCCTCAGCGGCGCGCGCAAGGAGGCCATCTCGGAGCGGACACAAGGTATCCTCACGAAACTGGCCGGCGCGGTGAGGATCATGGTCAGCGGCGCCATAGCCAAGATCCAAGGCGACTCACTGTTCCCGCACACGCCGAGCGCCGCAGGTGGCGTCCACCCGCTGACACGCAACGCCATGACCTGCGTCGAGCTGCTGGCGAGGCACCGCACCACGCTCGATCTGATCCTCGCGGGCGCCGACGAGCGCAGCTCCCTCGCC>marker3|348-348ATTATATCCCAAGTGAACCTAAAAGTTCTCCCACACAACGACGCAGGGTTTTACAAACAACCACCAAGGGCATGTTGTGGCGCGCCTGGGAGGGGTCCTTACAATTTCAACCTGACAGCGAAGTGCGGTGAGCCTGGTGCCTCGGCTTGCGCTGATCCTAAGACGCACTGGAGCTGGGATGGTATTCACTTGACAGAAGCCGCGTATCTGCACATTGCCAGAGGCTGGCTTCATGGACCGTTCGCAGACCAGCCTGTTGTACAAGCATCTTGAGTAATGTTGTGGCGCGCCTGGGAGGGGTCCTTACAATTTCAACCTGACAGCGAAGTGCGGTGAGCCTGGTGCCTCGGCTTGCGCTGATCCTAAGACGCACTGGAGCTGGGATGGTATTCACTTGACAGAAGCCGCGTATCTGCACATTGCCAGAGGCTGGCTTCATGGACCGTTCGCAGACCAGCCTGTTGTACAAGAGGGCATGTTGTGGCGCGCCTGGGAGGGGTCCTTACAATTTCAACCTGACAGCGAAGTGCGGTGAGCCTGGTGCCTCGGCTTGCGCTGATCCTAAGACGCACTGGAGCTGGGATGGTATTCACTTGATGAGTACAGTACACCCAGCCTCTCCTCCCTGCTGTTCTTGCACCTCTAGAGTGAGTGGCCACAATAGCACACACCGTGCGTGACACCAAACTGGATCCGATAAGGTANote
If there is a fasta sequence that does not conform to the agreed format, the primer design result for this sequence will not be output, and it will jump to the next sequence to try to design a primer.
Default settings
Default parameters include minimum, optimum, and maximum values for primer length, PCR product length, and Tm value. All sequences in the fasta file submitted at the same time use this set of parameters to design primers in batches. In the design process, these 9 parameters will be used for sequence design of conventional primers, and specific sequences will be added to the returned primer sequences.
Result
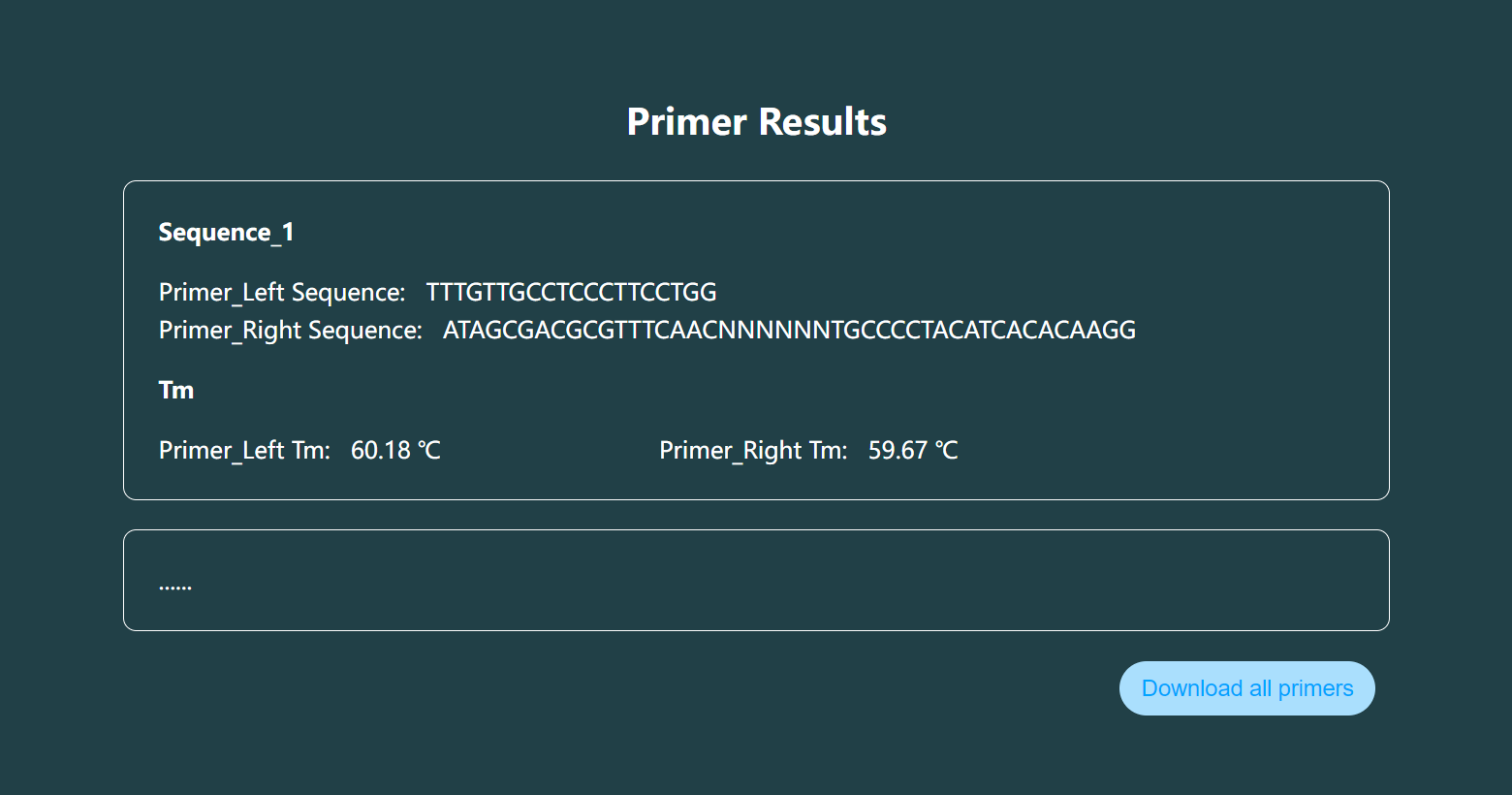
The result page only presents the primer design results of the first sequence on the page, and you need to download all the primer design results by clicking the "Download all primers" button at the bottom of the results page.
You need to click the "Download all primers" button at the bottom of the results page to download all the primer design results.

Header
Marker Name, Left Primer Sequence, Right Primer Sequence, Left Primer 5' Start Site, Right Primer 5' Start Site, Left Primer Length, Right Primer Length, Left Primer Tm Value, Right Primer Tm Value, Left Primer Primer GC Content, Right Primer GC Content, Product Length
iBP-seq
According to different application directions, iBP-seq can be divided into iBP-genotyping method for genotyping and iBP-epigenotyping method for methylation level detection.
Genotyping
Submit a task
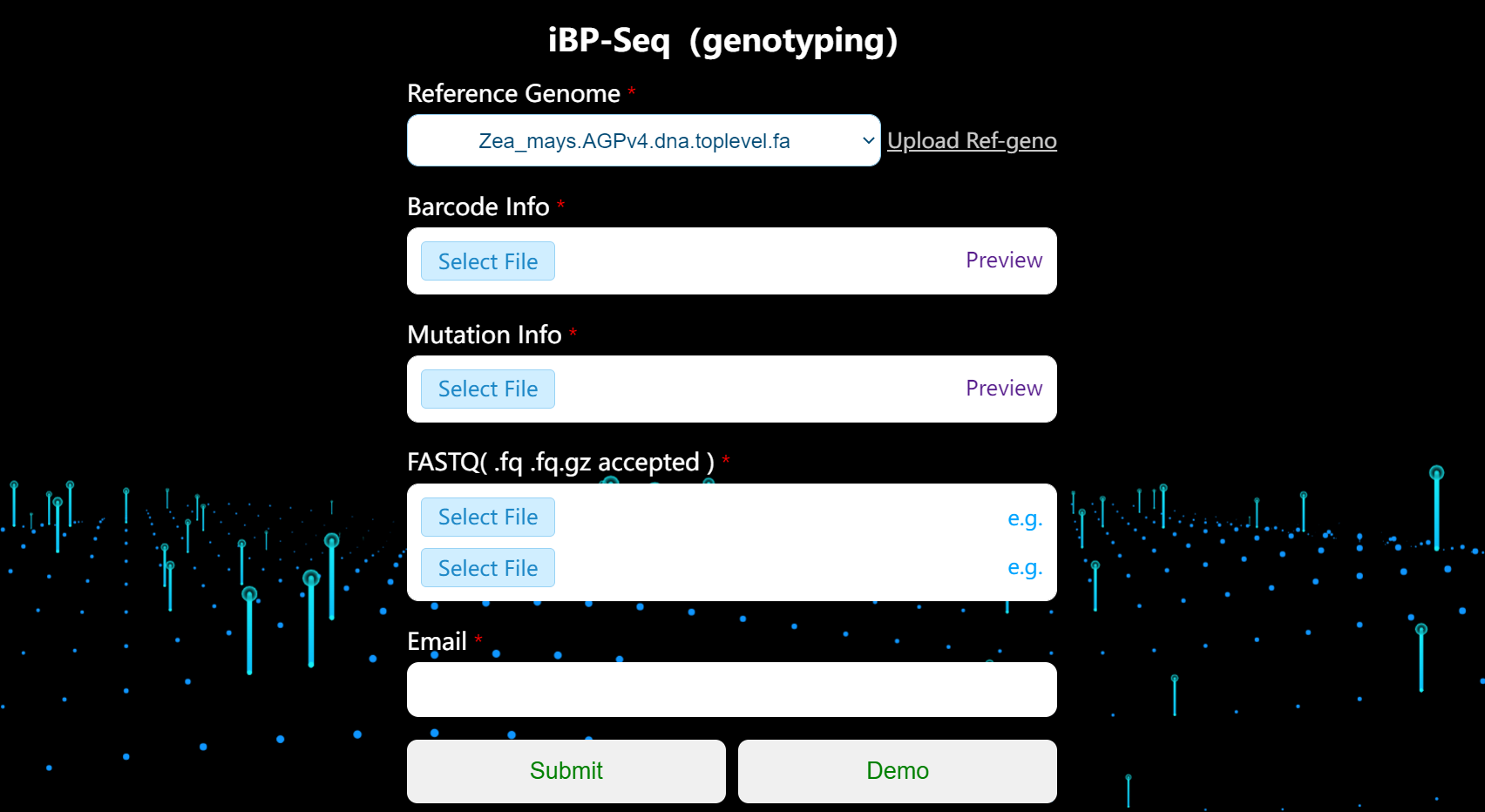
You can directly click the demo button on the submit page to run the preset example. Of course, you can also submit the following information required for iBP-genotyping analysis yourself.
[ optional ] Reference genome
You can select an existing reference genome from the drop-down box, or upload reference sequence by yourself.
[ required ] Barcode info
The barcode sequences you used in your experiment, they are all 8bp in length. Each line of barcode sequence corresponds to one sample.
[ required ] Mutation info
Variation information for molecular markers (SNP or InDel).
Data Format
CHROM Pos REF ALT 7 131102379 C G 3 14591067 C CGGAGGA zong31 28 GCCG GCCGGA zong31 33 ATGCACCG A Note
Here zong31 is the name of the custom reference sequence, this example is just to show the variety of optional mutation information formats, including SNP, Insert, Deletion and complex mutations, can be used for genotyping detection.
[ required ] Fastq file
Paired-end fastq files generated by next-generation sequencing. We prefer to upload files in .gz compressed format, which will further save your upload time.
[ required ] Email address
A valid email address to receive the analysis result.
Once the file upload is complete, you can leave the website and wait for an email notification of the result.
Result
The PNG image file helps you observe the genotyping results and efficiency from a macro perspective, and the xlsx file tells you the detailed information of each genotype.
PNG
Each picture corresponds to the genotype results of a population, and the title of the picture is also marked in the format (chromosome_locus_REF_ALT, the same as the content of the variation information file) for easy distinction. Each rug on the x-axis represents a sample, and its corresponding x-axis value represents the proportion of the mutated base at this site in the sequencing data (in short,
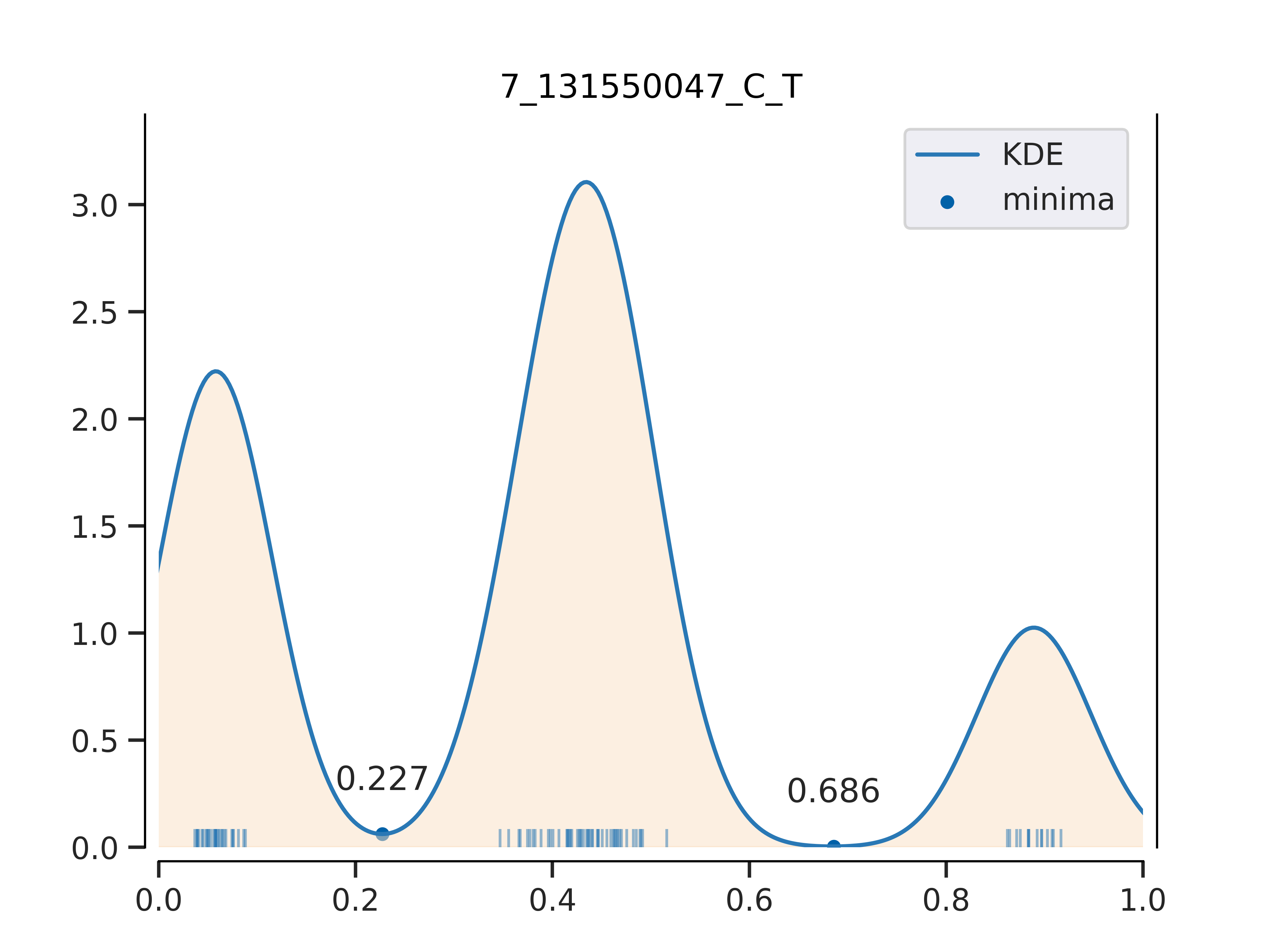 Fig.5 Kernel density distribution of mutation base frequency in population samples (chr7; 131550047bp; Ref=C; Alt=T)
Fig.5 Kernel density distribution of mutation base frequency in population samples (chr7; 131550047bp; Ref=C; Alt=T)XLSX
The first column corresponds to a sample for each num in order. The second column corresponds to the allele frequency of the ALT base at that locus for each sample. The third column 0, 1, and 2 represent three different genotypes, corresponding to the three peaks from left to right on the KDE curve.
Data Format
Num Frequency Genotype ... ... ... 6 0.057692 0 7 0.909091 2 8 0.45045 1 ... ... ... Header
Sample ID, Frequency of Alt Genotype, Sample Genotype
Epigenotyping
Submit a task
You can directly click the demo button on this page to run the preset example. Of course, you can also submit the following information required for iBP-epigenotyping analysis.
[ required ] Barcode info
Each row consists of the 8bp barcode sequence and the corresponding reference sequence name.
Data Format ATCACGTT,B73/Mo17 CGATGTTT,B73 TTAGGCAT,Mo17 TGACCACT,W22 ACAGTGGT,SK GCCAATGT,B73/Mo17/SK/W22 TGGTTGTT,B73/Mo17 ...
Format Description
barcode sequence, reference sequence 1/reference sequence 2/... (barcode sequence and reference sequence are separated by
","and multiple reference sequences are separated by"/")[ required ] Reference genome
You need to submit a fasta file containing multiple reference sequences for alignment.
Data Format
>1-B73 ATCCACGTCGCCAGAAATTCTTCACGCCGCTAGATTGTAAATCAATGGCTTCTATCCATCGTTGTATCGGTGGCGTGTGGAATGTCTCTTCTCACTCCTTCCGCTTCTCGAGACTTCCATGGTTGGCCCGTGCAGTGCATGCCTCTCCTCGCTCCCTTCGTCTCTCTGCCAGCTGCTTCACCTTCTGCCGCTTTGATCGTCCACTTCACTCCAGCACAGTCGCGTCGTCTACTGTCGCCTTCATCGTCTGCTTCGATGTTTGGTGATATCCATCACTTTCTTCATCGGCTCCACTTCCTCGTCGAGTTGCTGTTTCTTTCATTGGGGTTTCGAAGGGTGTGTTTGGTTGGATGTACATGGAGGGATGGAATAAGGTGGCCACATTTTCAATAGTGTTTGGA
>1-Mo17ATCCACGTCGCCAGAAATTCTTCACGCCGCTAGATTGTAAATCAATGGCTTCTATCCATCGTTGTATCGGTGGCGTGTGGAATGTCTCTTCTCACTCCTTCCGCTTCTCGAGACTTCCATGGTTGGCCCGTGCAGTGCATGCCTCTCCTCGCTCCCTTCGTCTCTCTGCCAGCTGCTTCACCTTCTGCCGCTTTGATCGTCCACTTCACTCCAGCACAGTCGCGTCGTCTACTGTCGCCTTCATCGTCTGCTTCGATGTTTGGTGATATCCATCGCTTTCTTCATCGGCTCCACTTCCTCGTCGAGTTGCTGTTTCTTTCATTGGGGTTTCGAAGGGTGTGTTTGGTTGGATGTACATGGAGGGATGGAATAAGGTGGCCACATTTTCAATAGTGTTTGGA>1-SKATCCACGTCGCCAGAAATTCTTCACACCGCTAGATTGTAAATCAATGACTTCTATCCATTGTTGTATCGGTGGCGTGTGGAATGTCTCTTCCCGCTCCTTCCACTTCTCGAGACTTCCATGGTTGGCCCATGCAGTGCATGCCTCTCCTCGCTCCCTTCGTCTCTCTGCCAGCTGCTTCACCTTCTGCCGCTTTGATCGTCCACTTCACTTCAGCACAGTCGCGTCGTCTACTGTCGCCTTCATCGTGTGCTTCGATGTTTGGTGATATCCATCGCTTTCTTCATCGGCTCCACTTCCTCGTCGAGTTGCTGCTTCTTTCATTGGGGTTTCGAAGGGTGTGTTTGGTTGGATGTACATGGAGGGATGAAATAACGTGGCCACATTTTTAATAGTGTTTGGA ...[ required ] Fastq file
Paired-end fastq files generated by next-generation sequencing. We prefer to upload files in .gz compressed format, which will further save your upload time. You can also download a pair of complete demos from the website.
[ required ] Email address
A valid email address to receive the analysis result.
Once the file upload is complete, you can leave the site and wait for an email notification of the result.
Result
PNG image files help you observe the results and efficiency of epigenotyping from a macro perspective, and csv files tell you the detailed information of each individual genotype.
PNG
Results of three different methylation types corresponding to several reference sequences.
 Fig.7 Visualization results of different methylation types
Fig.7 Visualization results of different methylation typesCSV
Detailed information on the type of methylation at the corresponding position of each reference sequence.
Data Format
chr pos context C_count CT_count ratio 1-B73 68 CG 2662 3965 0.671375 1-B73 86 CHH 714 3969 0.179894 1-B73 128 CHG 1814 3969 0.457042 Header
Chromosome Name, Position, Methylation Type, The Number of C in NGS Reads, The Total Number of C and T in NGS Reads, The Ratio of the Number of C in NGS Reads to the Total Number of C and T
Copyright
Copyright (c) Li Lab of HZAU, All rights reserved.